 贺巍巍,杨杰,王雨昕,李雨哲,陈达炜,赵云峰,周爽,方从容.超高效液相色谱-串联质谱法测定食品中高氯酸盐[J].中国食品卫生杂志,2017,29(4):438-444.
贺巍巍,杨杰,王雨昕,李雨哲,陈达炜,赵云峰,周爽,方从容.超高效液相色谱-串联质谱法测定食品中高氯酸盐[J].中国食品卫生杂志,2017,29(4):438-444.DOi:10.13590/j.cjfh.2017.04.010
超高效液相色谱-串联质谱法测定食品中高氯酸盐
(国家食品安全风险评估中心 卫生部食品安全风险评估重点实验室,北京100021)
作者简介: 贺巍巍女助理研究员研究方向为食品安全风险监测E-mail:heweiwei@cfsa.net.cn
通信作者: 方从容女主任技师研究方向为食品理化检验E-mail:fangcr@cfsa.net.cn
收稿日期: 2017-05-04
基金项目: 国家自然科学基金(21537001);国家食品安全风险评估中心高层次人才队伍建设523项目(1311613106702)
摘要:目的建立食品中高氯酸盐的超高效液相色谱-串联质谱(UPLC-MS/MS)的测定方法。方法香辛料调味品用水提取,蔬菜、水果用乙腈-水(1∶1,V/V)提取,肉、禽、蛋、奶和水产品用乙腈-水(2∶1,V/V)提取,C18柱(3 ml,200 mg)净化,以高氯酸根为内标定量,采用UPLC-MS/MS法测定。结果在0.3~20.0 μg/L范围内,3种提取溶液中高氯酸盐有良好的线性关系,R2≥0.999。加标浓度在2.0~50.0 μg/kg范围内,内标相对平均回收率为82.6%~108.6%,相对标准偏差(RSD)为1.0%~9.9%。牛奶的定量限为2.0 μg/kg,其他食品的定量限为10.0 μg/kg。结论本方法简单快捷,定量准确,适用于食品中高氯酸盐的测定。
关键词:
超高效液相色谱-串联质谱法; 高氯酸盐; 食品污染物; 食品安全; 检测方法
中图分类号: R155 文献标识码:A 文章编号:1004-8456(2017)04-0438-07
Determination of perchlorate in food by ultra performance liquid
chromatography-tandem mass spectrometry
chromatography-tandem mass spectrometry
(Key Laboratory of Food Safety Risk Assessment of Ministry of Health,China National Center for Food Safety Risk Assessment,Beijing 100021,China)
Abstract:ObjectiveTo establish a method for the determination of perchlorate in food by ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS). MethodsThe perchlorate residue in spices and condiments was extracted with water, that in vegetables and fruits was extracted with acetonitrile-water (1∶ 1, V/V), and that in meat, poultry, eggs, milk and aquatic products was extracted with acetonitrile-water (2∶ 1, V/V). The supernatant was cleaned up with C18 SPE (3 ml, 200 mg), and the detection was carried out by UPLC-MS/MS with internal standardmethod for quantification. ResultsThe calibration curve was linear in the concentration range of 0.3-20.0 μg/L (R2≥0.999), the recovery was in the range of 82.6%-108.6%, the relative standard deviation (RSD) was in the range of 1.0%-9.9%, and the limit of detection was 2.0 μg/kg for milk, and 10.0 μg/kg for other food. ConclusionThe method was simple, accurate and highly sensitive, and suitable for the determination of perchlorate in food.
Key words:
Ultra performance liquid chromatography-tandem mass spectrometry; perchlorate; food contaminants; food safety; testing methods
高氯酸盐为国际上广泛关注的环境污染物。2014年欧盟食品安全局(EFSA)对食品中高氯酸盐的健康危害进行了再评估。EFSA根据收集的水果、蔬菜及其制品中高氯酸盐的污染资料及文献中果汁、酒精饮料、牛奶、婴儿配方食品及母乳中高氯酸盐含量评估了高氯酸盐的毒性和暴露量,并根据对健康成年人甲状腺碘的抑制作用,建立了高氯酸盐耐受摄入量(TDI)为0.3 g/kg BW,同时,EFSA认为高氯酸盐对胎儿和婴儿具有潜在的急性影响。根据已获得的食品和饮用水中高氯酸盐的含量,EFSA认为单次暴露高氯酸盐不可能造成健康危害,因此,尚没有必要建立高氯酸盐的急性参考剂量。但是,长期暴露高氯酸盐对人体的健康危害值得关注,尤其是对于患有轻中度碘缺乏的年轻人群,特别是对于碘摄入量低的哺乳婴儿和低年龄儿童更应关注高氯酸盐的短期暴露[1]。在EFSA评估报告中, 高氯酸盐浓度在蔬菜中的加权范围值为4.8~111 μg/kg,水果加权范围值为0.5~28 μg/kg,蔬菜和水果汁加权范围值为0.047~463.5 μg/kg,婴幼儿配方粉加权平均值为10 μg/kg,牛奶加权平均值为6.8 μg/kg,全麦面粉加权平均值为3.5 μg/kg,大米加权平均值为1 μg/kg,鱼和渔业产品加权范围值为0.32~1 593 μg/kg[1]。ZHANG等[2]对中国7个城市的灰尘、人尿液、饮用水、乳制品中的高氯酸盐浓度进行了检测,高氯酸盐含量为灰尘1.67~821 μg/g 干重,检出率为100%;人尿液<1.00~995 ng/ml,检出率为99%;饮用水0.26~280 ng/ml,检出率为100%;乳制品1.74~22.0 ng/ml,检出率为100%,为此,广泛了解各类食品中高氯酸盐的污染水平极为必要。
食品中高氯酸盐的测定主要有离子色谱(IC)法[3-5]、离子色谱-串联质谱(IC-MS/MS)法[6-11]、液相色谱-串联质谱(LC-MS/MS)法[12-15]等。为提高食品中高氯酸盐检测的灵敏度,美国环保局(EPA)于2007年提出了IC-MS/MS用于高氯酸盐测定。但在实际应用中发现IC-MS/MS在国内存在普及率低、离子色谱柱不耐受有机溶剂、大体积进样易造成柱子过载等问题。随着LC-MS/MS性能和检测灵敏度的提高,其在高氯酸盐检测中的应用日渐增加。我国SN/T 4089—2015《进出口食品中高氯酸盐的测定 液相色谱-质谱/质谱法》[15]建立了采用阴离子色谱柱进行高氯酸盐测定的液相色谱-串联质谱法。通过方法验证发现,高氯酸盐液相色谱峰型宽(保留时间的跨度约1 min),且流动相含有高浓度的盐溶液,易对质谱仪造成污染。为提高食品中高氯酸盐测定的准确性,本试验在原有工作基础上,扩大了食品范围,优化了前处理方法和仪器方法,进一步验证了超高效液相色谱-串联质谱(UPLC-MS/MS)法测定不同食品基质中高氯酸盐的应用性。
乙腈、甲醇、甲酸均为色谱纯,高氯酸根标准溶液(ClO4-,浓度1 000 mg/L,美国INORGANIC),高氯酸根内标溶液(Cl18O4-,浓度100 mg/L,美国Cambridge Isotope Laboratories),提取溶液1为乙腈-水(1∶1,V/V),提取溶液2为乙腈-水(2∶1,V/V),超纯水(电导率<1.0 μs/cm,由Milli-Q超纯水系统制得),SynergiTM Max-RP柱(4.6 mm×150 mm,4 μm,美国phenom-enex),Sep-pak Vac C18(3 ml,200 mg,美国Waters)。
牛奶、豆浆、含乳饮料:称取样品5 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加水5 ml,混匀,加20 ml乙腈,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。
谷物和动物源性食品:称取样品5 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加20 ml提取溶液2,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。
香辛料调味品(桂皮、花椒、大料、五香粉、咖喱粉等):称取样品1 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加20 ml水,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。
质谱:电喷雾离子源(ESI),负离子模式;毛细管电压4 kV;雾化气流速3 L/min;干燥气流速10 L/min;加热气流速10 L/min;脱溶剂气温度300 ℃;接口温度350 ℃;加热模块温度400 ℃;碰撞器压力270 kPa;多反应监测(MRM)离子对:高氯酸m/z 99.1/83.0(定量离子)、101.1/85.0,高氯酸内标m/z 107.1/88.9(定量离子);离子对碰撞能均
为-26 V。
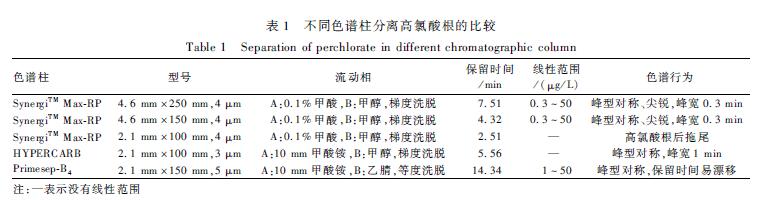
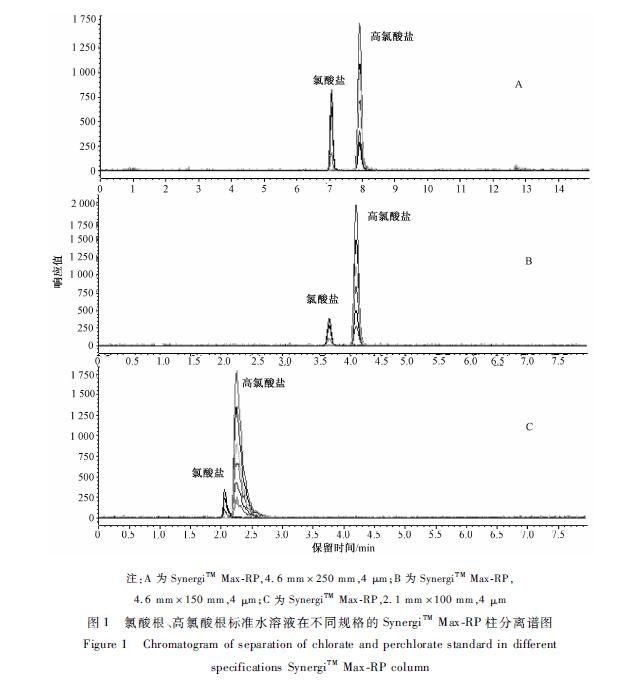
在确定SynergiTM Max-RP柱为分析柱后,又对3种规格的SynergiTM Max-RP柱(4.6 mm×250 mm,4 μm、 4.6 mm×150 mm,4 μm和2.1 mm×100 mm,4 μm)的性能进行优化,并从高氯酸根保留时间、峰型和灵敏度3个方面考察其色谱柱的性能。结果显示:从峰型、灵敏度考虑,250 mm柱=150 mm柱>100 mm柱;250 mm柱保留时间为7.51 min,完成全部分析需要18 min;150 mm柱保留时间为4.32 min,完成全部分析需要11 min。250 mm柱和150 mm柱的峰型和灵敏度都能满足分析的要求,因此综合考虑后采用SynergiTM Max-RP(4.6 mm×150 mm,4 μm)为分析柱。不同色谱柱分离高氯酸根的比较见表1,不同规格的SynergiTM Max-RP柱分离高氯酸根和氯酸根的情况见图1。
应用乙腈-水体系(其中乙腈与水的比例为0∶1、1∶1、2∶1,V/V)对上述13种样品中的高氯酸盐进行提取,C18 SPE柱净化。从色谱行为看,水果、蔬菜类样品中的香蕉、油菜采用水提取,进样液中的高氯酸根色谱峰型变宽,峰分叉,且C18 SPE净化液略有浑浊;采用乙腈-水(1∶1,V/V)提取,香蕉、油菜进样液中的高氯酸根色谱峰型明显改善,峰型对称,无拖尾,且C18 SPE净化液澄清,因此水果、蔬菜类的提取溶剂为乙腈-水(1∶1,V/V)。香辛料调味品采用水为提取溶剂时,提取液颜色浅,提取液中含有的色素、脂肪酸、芳香类化合物等杂质明显少于采用含有乙腈的提取溶剂得到的提取液,且进样液中的高氯酸根色谱峰型对称、尖锐,高氯酸根内标绝对回收率在40%~100%之间,因此将水作为香辛料调味品的提取溶剂。乙腈具有很好地沉淀蛋白的作用,经过比较,乙腈和水体积比例为2∶1对沉淀面粉、牛奶、鸡蛋、牛肉、桂鱼中的蛋白质的效果最好,因此谷物和动物源性食品的提取溶剂为乙腈-水(2∶1,V/V)。
文献[6]和SN/T 4089—2015[15]中均采用乙腈-1%乙酸体系作为提取溶剂,而本试验采用乙腈-水体系作为提取溶剂,因此需考察两种提取溶剂体系对不同食品基质中高氯酸盐的提取率。分别按SN/ T 4089—2015和本试验的提取溶剂体系、提取步骤对麻椒(10.30 mg/kg)、散玉米面(0.54 mg/kg)、有机小米(0.063 mg/kg)、油菜(0.26 mg/kg)、菠萝(0.040 mg/kg)和草鱼(0.050 mg/kg)6种样品中的高氯酸盐进行提取,并将提取液用水进行稀释,以消除液质测定中基质效应的影响。其中,麻椒提取液稀释100倍,散玉米面提取液稀释20倍,其他4种样品提取液分别稀释10倍,经4 ℃ 10 000 r/min离心10 min后,取上清液进行UPLC-MS/MS分析,并用外标法定量。结果表明,本试验方法和SN/T 4089—2015方法测定的6种食品基质中高氯酸盐含量差异无统计学意义(P>0.05),且两种方法样品中高氯酸盐含量相对标准偏差在3.6%~7.1%之间,因此,本试验采用乙腈-水体系,并通过涡旋和超声提取的方式,完全可以将不同基质食品中的高氯酸盐提取出来。
的检出限,即牛奶中高氯酸盐的检出限为0.6 μg/kg,蔬菜、水果、谷物、动物源性食品、香辛料调味品为1.8 μg/kg。
食品中高氯酸盐的测定主要有离子色谱(IC)法[3-5]、离子色谱-串联质谱(IC-MS/MS)法[6-11]、液相色谱-串联质谱(LC-MS/MS)法[12-15]等。为提高食品中高氯酸盐检测的灵敏度,美国环保局(EPA)于2007年提出了IC-MS/MS用于高氯酸盐测定。但在实际应用中发现IC-MS/MS在国内存在普及率低、离子色谱柱不耐受有机溶剂、大体积进样易造成柱子过载等问题。随着LC-MS/MS性能和检测灵敏度的提高,其在高氯酸盐检测中的应用日渐增加。我国SN/T 4089—2015《进出口食品中高氯酸盐的测定 液相色谱-质谱/质谱法》[15]建立了采用阴离子色谱柱进行高氯酸盐测定的液相色谱-串联质谱法。通过方法验证发现,高氯酸盐液相色谱峰型宽(保留时间的跨度约1 min),且流动相含有高浓度的盐溶液,易对质谱仪造成污染。为提高食品中高氯酸盐测定的准确性,本试验在原有工作基础上,扩大了食品范围,优化了前处理方法和仪器方法,进一步验证了超高效液相色谱-串联质谱(UPLC-MS/MS)法测定不同食品基质中高氯酸盐的应用性。
1材料与方法
1.1主要仪器与试剂
8050高效液相色谱-质谱/质谱仪(配备电喷雾离子源ESI,日本岛津)、固相萃取(SPE)装置、冷冻离心机(最大转速10 000 r/min)。乙腈、甲醇、甲酸均为色谱纯,高氯酸根标准溶液(ClO4-,浓度1 000 mg/L,美国INORGANIC),高氯酸根内标溶液(Cl18O4-,浓度100 mg/L,美国Cambridge Isotope Laboratories),提取溶液1为乙腈-水(1∶1,V/V),提取溶液2为乙腈-水(2∶1,V/V),超纯水(电导率<1.0 μs/cm,由Milli-Q超纯水系统制得),SynergiTM Max-RP柱(4.6 mm×150 mm,4 μm,美国phenom-enex),Sep-pak Vac C18(3 ml,200 mg,美国Waters)。
1.2方法
1.2.1标准溶液的配制
高氯酸根标准使用液(1.0 mg/L):吸取高氯酸根标准溶液,用水配制成中间液(10 mg/L);吸取中间液1.0 ml,置10 ml容量瓶中,用水稀释至刻度,混匀,置4 ℃冰箱中保存。高氯酸根内标使用液(1.0 mg/L):吸取高氯酸根内标溶液,用水配制成中间液(10 mg/L);吸取中间液1.0 ml,置10 ml容量瓶中,用水稀释至刻度,混匀,置4 ℃冰箱中保存。高氯酸根系列标准液:使用前,分别吸取高氯酸根标准使用液和内标使用液适量,分别用水、提取溶液1、提取溶液2稀释,制成高氯酸根浓度分别为0.3、0.5、1.0、2.0、10.0、20.0 μg/L的系列标准液,各溶液中内标高氯酸根的浓度为 5.0 μg/L。
1.2.2样品前处理
水果、蔬菜:称取样品5 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加20 ml提取溶液1,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。牛奶、豆浆、含乳饮料:称取样品5 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加水5 ml,混匀,加20 ml乙腈,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。
谷物和动物源性食品:称取样品5 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加20 ml提取溶液2,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。
香辛料调味品(桂皮、花椒、大料、五香粉、咖喱粉等):称取样品1 g(精确到0.01 g)于50 ml离心管中,加高氯酸根内标使用液(1.0 mg/L)150 μl,加20 ml水,1 600 r/min涡旋混匀5 min,超声提取20 min后,4 ℃ 10 000 r/min离心10 min,上清液转移至另一支50 ml离心管中,用水稀释至30 ml,混匀,待净化。
1.2.3净化
C18柱在使用前依次用3 ml乙腈、3 ml水溶液活化,重力条件下自然流出。吸取1.2.2中上清液2.0 ml过C18 SPE柱,弃去前1.0 ml流出液,收集流出液1.0 ml,涡旋10 s,供UPLC-MS/MS测定。
1.2.4仪器条件
色谱:SynergiTM MAX-RP色谱柱(4.6 mm×150 mm,4 μm);流速0.5 ml/min;流动相A:0.1%甲酸溶液,流动相B:甲醇;梯度洗脱:10%B保持1 min,在1 min内增至15%B,保持3 min,再在0.1 min内增至95%B,保持3 min;柱温30 ℃;进样量10 μl。质谱:电喷雾离子源(ESI),负离子模式;毛细管电压4 kV;雾化气流速3 L/min;干燥气流速10 L/min;加热气流速10 L/min;脱溶剂气温度300 ℃;接口温度350 ℃;加热模块温度400 ℃;碰撞器压力270 kPa;多反应监测(MRM)离子对:高氯酸m/z 99.1/83.0(定量离子)、101.1/85.0,高氯酸内标m/z 107.1/88.9(定量离子);离子对碰撞能均
为-26 V。
2结果与分析
2.1色谱柱的选择
高氯酸根在C18柱上难以保留,因此选用了SynergiTM Max-RP柱(4.6 mm×250 mm,4 μm)、HYPERCARB柱(2.1 mm×100 mm,3 μm)和Primesep-B4(2.1 mm×150 mm,5 μm)为分析柱,流动相为0.1%甲酸-甲醇体系,比较50 μg/L高氯酸根标准水溶液在不同色谱柱上的色谱行为。结果表明,高氯酸根在Primesep-B4柱上无信号,更换流动相为甲酸铵-乙腈体系,高氯酸根保留时间为14.34 min,但灵敏度低且保留时间易漂移;在SynergiTMMax-RP柱和HYPERCARB柱上,高氯酸根峰型对称,与氯酸根的分离度都>1.5,但在同种浓度下,HYPERCARB柱的高氯酸根响应值比SynergiTM Max-RP柱的高氯酸根响应值低10倍,因此,选择SynergiTM Max-RP柱为分析柱。在确定SynergiTM Max-RP柱为分析柱后,又对3种规格的SynergiTM Max-RP柱(4.6 mm×250 mm,4 μm、 4.6 mm×150 mm,4 μm和2.1 mm×100 mm,4 μm)的性能进行优化,并从高氯酸根保留时间、峰型和灵敏度3个方面考察其色谱柱的性能。结果显示:从峰型、灵敏度考虑,250 mm柱=150 mm柱>100 mm柱;250 mm柱保留时间为7.51 min,完成全部分析需要18 min;150 mm柱保留时间为4.32 min,完成全部分析需要11 min。250 mm柱和150 mm柱的峰型和灵敏度都能满足分析的要求,因此综合考虑后采用SynergiTM Max-RP(4.6 mm×150 mm,4 μm)为分析柱。不同色谱柱分离高氯酸根的比较见表1,不同规格的SynergiTM Max-RP柱分离高氯酸根和氯酸根的情况见图1。
|
表1不同色谱柱分离高氯酸根的比较 Table 1Separation of perchlorate in different chromatographic column 注:—表示没有线性范围 |
|
注:A为SynergiTM Max-RP,4.6 mm×250 mm,4 μm;B为SynergiTM Max-RP, 4.6 mm×150 mm,4 μm;C为SynergiTM Max-RP,2.1 mm×100 mm,4 μm 图1氯酸根、高氯酸根标准水溶液在不同规格的SynergiTM Max-RP柱分离谱图 Figure 1Chromatogram of separation of chlorate and perchlorate standard in different specifications SynergiTM Max-RP column |
2.2提取溶剂的优化
常见的高氯酸盐包括高氯酸铵、高氯酸钠、高氯酸锂、高氯酸镁、高氯酸银、高氯酸钾、高氯酸铯等。根据高氯酸盐中除高氯酸钾和高氯酸铯可溶于水外,其他都易溶于水的化学性质,参考SN/T 4089—2015[15]的方法,选择乙腈-水体系、乙腈-0.2%乙酸体系和乙腈-1%乙酸体系为提取溶剂,但在前期试验中发现,用含有1%乙酸溶液配制的高氯酸根标准溶液,在SynergiTM Max-RP色谱柱上,其高氯酸根色谱峰为双峰,而用乙腈-水体系和乙腈-0.2%乙酸体系配制的高氯酸根标准溶液,在SynergiTM Max-RP色谱柱为单峰,因此初步选择提取溶剂为乙腈-水体系和乙腈-0.2%乙酸体系(其中乙腈与水相的比例为0∶1、1∶1、2∶1,V/V),分别对水果(梨、香蕉)、蔬菜(西红柿、油菜、藕、蘑菇)、香辛料调味品(五香粉、花椒)、谷物(面粉)、动物源性食品(牛奶、鸡蛋、牛肉、桂鱼)13种样品进行提取,并经过C18 SPE柱净化后,比较同一食品在不同提取溶剂中的色谱行为及基质效应。试验结果显示经乙腈-0.2%乙酸体系提取的水果、蔬菜、香辛料调味品、面粉样品的内标绝对回收率明显高于乙腈-水体系,且内标绝对回收率在200%~300%,这是由于上述样品中溶于酸和乙腈的物质造成了基质效应的增加。而经乙腈-水体系和乙腈-0.2%乙酸体系分别提取的牛奶、鸡蛋、牛肉、桂鱼样品中的内标绝对回收率结果相当(80%~120%)。采用含酸溶液提取的目的主要是改善峰型和提高提取率,但由于高氯酸盐大多易溶于水,在提取溶液中加入酸的意义不大,因此舍去乙腈-0.2%乙酸体系,选用乙腈-水体系为提取溶剂。应用乙腈-水体系(其中乙腈与水的比例为0∶1、1∶1、2∶1,V/V)对上述13种样品中的高氯酸盐进行提取,C18 SPE柱净化。从色谱行为看,水果、蔬菜类样品中的香蕉、油菜采用水提取,进样液中的高氯酸根色谱峰型变宽,峰分叉,且C18 SPE净化液略有浑浊;采用乙腈-水(1∶1,V/V)提取,香蕉、油菜进样液中的高氯酸根色谱峰型明显改善,峰型对称,无拖尾,且C18 SPE净化液澄清,因此水果、蔬菜类的提取溶剂为乙腈-水(1∶1,V/V)。香辛料调味品采用水为提取溶剂时,提取液颜色浅,提取液中含有的色素、脂肪酸、芳香类化合物等杂质明显少于采用含有乙腈的提取溶剂得到的提取液,且进样液中的高氯酸根色谱峰型对称、尖锐,高氯酸根内标绝对回收率在40%~100%之间,因此将水作为香辛料调味品的提取溶剂。乙腈具有很好地沉淀蛋白的作用,经过比较,乙腈和水体积比例为2∶1对沉淀面粉、牛奶、鸡蛋、牛肉、桂鱼中的蛋白质的效果最好,因此谷物和动物源性食品的提取溶剂为乙腈-水(2∶1,V/V)。
文献[6]和SN/T 4089—2015[15]中均采用乙腈-1%乙酸体系作为提取溶剂,而本试验采用乙腈-水体系作为提取溶剂,因此需考察两种提取溶剂体系对不同食品基质中高氯酸盐的提取率。分别按SN/ T 4089—2015和本试验的提取溶剂体系、提取步骤对麻椒(10.30 mg/kg)、散玉米面(0.54 mg/kg)、有机小米(0.063 mg/kg)、油菜(0.26 mg/kg)、菠萝(0.040 mg/kg)和草鱼(0.050 mg/kg)6种样品中的高氯酸盐进行提取,并将提取液用水进行稀释,以消除液质测定中基质效应的影响。其中,麻椒提取液稀释100倍,散玉米面提取液稀释20倍,其他4种样品提取液分别稀释10倍,经4 ℃ 10 000 r/min离心10 min后,取上清液进行UPLC-MS/MS分析,并用外标法定量。结果表明,本试验方法和SN/T 4089—2015方法测定的6种食品基质中高氯酸盐含量差异无统计学意义(P>0.05),且两种方法样品中高氯酸盐含量相对标准偏差在3.6%~7.1%之间,因此,本试验采用乙腈-水体系,并通过涡旋和超声提取的方式,完全可以将不同基质食品中的高氯酸盐提取出来。
2.3净化柱的选择
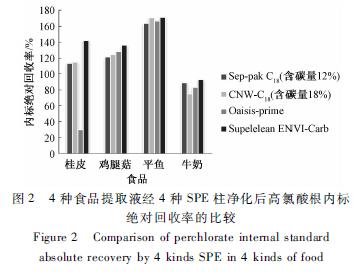
试验选择Sep-pack C18柱(200 mg,含碳量为12%)、CNW C18柱(200 mg,含碳量为18%)、Oasis-prime(200 mg)和Supelelean ENVI-Carb石墨碳黑柱(500 mg)4种SPE柱,并采用通过式净化方式。首先将只含有高氯酸根内标的水空白、提取溶液1空白、提取溶液2空白3种提取溶剂经4种SPE柱净化,以考察高氯酸根内标回收率情况。其次以鸡腿菇、牛奶、平鱼、桂皮为样品,按照1.2.2样品前处理方法进行提取,并分别经上述4种SPE柱净化,以比较4种SPE柱对上述样品中杂质的净化效果。试验结果显示,3种含高氯酸根内标的空白溶剂经Sep-pack C18柱、CNW C18柱、Oasis-prime净化后,高氯酸根内标绝对回收率均为100%,而Supelelean ENVI-Carb石墨碳黑柱对3种空白溶液中的高氯酸根内标均有极强的吸附,内标绝对回收率为0%,但在样品净化过程中是优先吸附样品提取液中的杂质,因此在同种样品净化试验中,Supelelean ENVI-Carb石墨碳黑柱的内标绝对回收率与其他3种SPE柱中的内标绝对回收率相当。桂皮样品的提取液经Oasis-prime SPE柱净化后,高氯酸根内标绝对回收率只有30%,其水空白中的内标回收率可达到100%,说明此SPE柱吸附杂质的效果差,且净化液中的杂质在质谱上产生了强抑制。4种样品提取液经含碳量为12%和18%的不同C18 SPE柱净化后,同种样品间的净化液颜色和高氯酸根内标绝对回收率差异无统计学意义(P>0.05)。综合考虑后,选择Sep-pack C18柱(200 mg,含碳量为12%)为净化柱。4种食品提取液经SPE柱净化后高氯酸根内标绝对回收率的比较见图2。
|
图24种食品提取液经4种SPE柱净化后高氯酸根内标 绝对回收率的比较 Figure 2Comparison of perchlorate internal standard absolute recovery by 4 kinds SPE in 4 kinds of food |
2.4基质效应
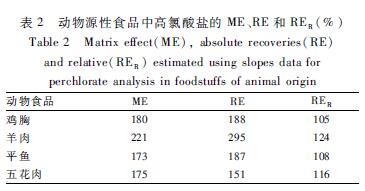
采用常用的标准曲线比较法[16]对建立的UPLC-MS/MS方法中的动物源性食品的前处理方法进行基质效应的评估。标准曲线A,用提取溶剂2配制标准曲线,浓度为0、2、5、10、20、50 μg/kg;标准曲线B,分别选择不含高氯酸盐的鸡胸、羊肉、平鱼和五花肉4种样品,按照1.2.2样品前处理方法进行提取、净化,并于上机前用净化液配制浓度范围为上述6个浓度点的标准曲线;标准曲线C,4个样品制备前,分别进行上述6个浓度点的加标,再按同样方法进行提取和净化。基质效应(ME,%)=斜率B曲线/斜率A曲线×100%,绝对回收率(RE,%)=斜率C曲线/斜率A曲线×100%,相对回收率(RER,%)=斜率C曲线/斜率B曲线×100%。结果显示,ME为173%~221%,RE为151%~295%,即采用本方法的4种样品基质效应为基质增强,因此如采用外标法定性定量误差极大,而采用同位素稀释法可有效的解决因基质效应造成定量不准确的问题。动物源性食品中高氯酸盐的ME、RE和RER结果见表2。
|
表2动物源性食品中高氯酸盐的ME、RE和RER(%) Table 2Matrix effect(ME), absolute recoveries(RE) and relative(RER) estimated using slopes data for perchlorate analysis in foodstuffs of animal origin |
2.5线性范围
用水、提取溶剂1和提取溶剂2分别制成高氯酸根浓度分别为0.30~20.0 μg/L的标准工作液,吸取10 μl系列标准溶液注入UPLC-MS/MS进行分析,内标法定量,得线性回归方程为y1=1.68224x+0.007166(水配制),R2=0.999 9;y2=1.61879x+0.02065(提取溶剂1),R2=0.999 9,y3=1.62676x+0.02926(提取溶剂2),R2=0.999 8。
2.6准确度和精密度
分别选取西红柿、油菜、香蕉、平鱼、牛肉、牛奶、大米、面粉、桂皮9种样品,按1.2.2样品前处理方法进行处理,牛奶中添加2、5、10 μg/kg的高氯酸盐,其他食品添加水平为10、20、50 μg/kg的高氯酸盐,回收率试验结果见表3。
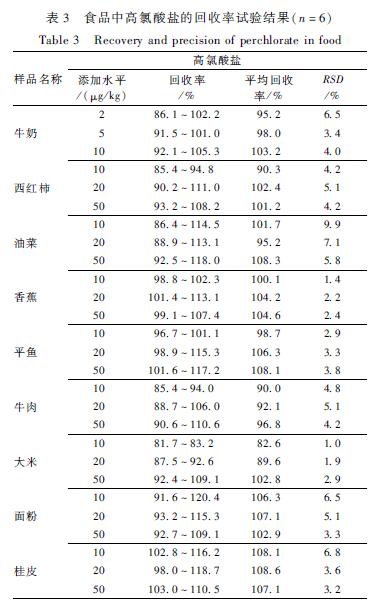
|
表3食品中高氯酸盐的回收率试验结果(n=6) Table 3Recovery and precision of perchlorate in food |
2.7定量限及检出限
根据回收率试验结果,低水平加标试验结果符合定量要求,以此作为方法的定量限,即牛奶中高氯酸盐的定量限为2.0 μg/kg,蔬菜、水果、谷物、动物源性食品、香辛料调味品为10.0 μg/kg;以信噪比为3的含量作为方法的检出限,即牛奶中高氯酸盐的检出限为0.6 μg/kg,蔬菜、水果、谷物、动物源性食品、香辛料调味品为1.8 μg/kg。
2.8样品测定
采用建立的方法测定购自北京市内超市和市场的163份样品,包括蔬菜81份、动物源性食品(肉、禽、蛋和水产品)28份、牛奶等饮品20份、谷物17份、香辛料调味品17份。目前,国际上尚没有高氯酸盐限量标准,欧盟2014年对高氯酸盐的评估报告[1]中,规定了欧盟成员国之间的高氯酸盐推荐限量值(见表4),据此浓度值,在17份谷物样品中有1份有机小米(0.063 mg/kg)、1份散玉米面(0.54 mg/kg)超过欧盟其他食品的推荐值0.05 mg/kg,17份香辛料调味品中有9份样品高氯酸盐含量超过了1.0 mg/kg,这9份样品全部为花椒,含量范围为1.10~10.30 mg/kg,蔬菜和动物源性食品均在欧盟推荐的浓度值内。163份样品中高氯酸盐含量范围见表5。
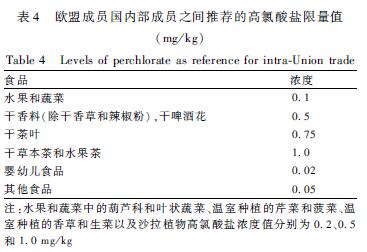
|
表4欧盟成员国内部成员之间推荐的高氯酸盐限量值(mg/kg) Table 4Levels of perchlorate as reference for intra-Union trade 注:水果和蔬菜中的葫芦科和叶状蔬菜、温室种植的芹菜和菠菜、温室种植的香草和生菜以及沙拉植物高氯酸盐浓度值分别为0.2、0.5和1.0 mg/kg |
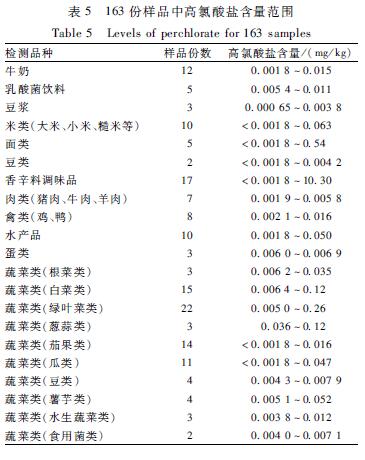
|
表5163份样品中高氯酸盐含量范围 Table 5Levels of perchlorate for 163 samples |
3小结
本方法选用SynergiTM Max-RP为分析柱,流动相体系为0.1%甲酸-甲醇溶液,C18 SPE柱净化,并采用UPLC-MS/MS进行分析和同位素内标法定量。此方法适用于食品中高氯酸盐的测定。
参考文献
[1]European Food Safety Authority.Scientific opinion on the risks to public health related to the presence of perchlorate in food,in particular fruits and vegetables[J].EFSA Journal,2014,12(10):3869.
[2]ZHANG T,CHEN X J,WANG D,et al.Perchlorate in indoor dust and human urine in China:contribution of indoor dust to total daily intake[J].Environmental Science and Technology,2015,49(4):2443-2450.
[3]李柚,喻宏伟,王飞.离子色谱法测定葡萄酒中的高氯酸盐[J].色谱,2016,34(10):989-911.
[4]孟维伟.茶叶中高氯酸盐的检测——离子色谱法[J].轻工科技,2016,216(11):12-13.
[5]丛鑫,苏葳艺,赵晓云,等.离子色谱法同时测定牛乳及牛乳制品中硫氰酸盐和高氯酸盐[J].沈阳医药科技大学学报,2013,30(8):601-604.
[6]U.S.Food and Drug Administration.Revision 2:rapid determination of perchlorate anion in foods by ion chromatography-tandem mass spectrometry [EB/OL].(2005-04-12) [2017-04-01].ttps://www.fda.gov/Food/FoodborneIllnessContaminants/ChemicalContaminan-ts/ucm077793.htm.
[7]SHI Y L,ZHANG P,WANG Y W,et al.Perchlorate in sewage sludge,rice,bottled water and milk collected from different areas in China[J].Science Direct,2007,33(7):955-962.
[8]ARIBI H E,BLANC Y J C L,ANTONSEN S,et al.Analysis of perchlorate in foods and beverages by ion chromatography coupled with tandem mass spectrometry (IC-ESI-MS/MS)[J].Analytica Chimica Acta,2006,567(1):39-47.
[9]KIRK A B,KROLL M,DYKE J V,et al.Perchlorate,iodine supplements,iodized salt and breast milk iodine content[J].Science of the Total Environment,2012,420(1):73-78.
[10]BLASINI L V,BLOUNT B C,SANTOS S O,et al.Perchlorate exposure and dose estimates in infants[J].Environ Sci Technol,2011,45(9):4127-4132.
[11]BORJAN M,MARCELLA S,BLOUNT B,et al.Perchlorate exposure in lactating women in an urban community in New Jersey[J].Science of the Total Environment,2011,409(3):460-464.
[12]LI Y T,GEORGE E J.Analysis of perchlorate in water by reversed phase LC/ESI-MS/MS using an internal standard technique[J].Analytical Chemistry,2005,77(14):4453-4458.
[13]PISARENKO A N,STANFORD B D,QUINONES O,et al.Rapid analysis of perchlorate,chlorate and bromate ions in concentrated sodium hypochlorite solutions[J].Analytica Chimica Acta,2010,659(1/2):216-223.
[14]黄晓兰,罗辉泰,吴惠勤,等.奶粉中高氯酸盐的液相色谱-串联质谱测定[J].分析测试学报,2009,28(8):896-899.
[15]中华人民共和国国家质量监督检验检疫总局.进出口食品中高氯酸盐的测定 液相色谱-质谱/质谱法:SN/T 4089—2015[S].北京:中国标准出版社,2015.
[16]HOFF R B,RBENSAM G,JANK L,et al.Analytical quality assurance in veterinary drug residue analysis methods:matrix effects determination and monitoring for sulfonamides analysis[J].Talanta,2015,132(6):443-450.
[2]ZHANG T,CHEN X J,WANG D,et al.Perchlorate in indoor dust and human urine in China:contribution of indoor dust to total daily intake[J].Environmental Science and Technology,2015,49(4):2443-2450.
[3]李柚,喻宏伟,王飞.离子色谱法测定葡萄酒中的高氯酸盐[J].色谱,2016,34(10):989-911.
[4]孟维伟.茶叶中高氯酸盐的检测——离子色谱法[J].轻工科技,2016,216(11):12-13.
[5]丛鑫,苏葳艺,赵晓云,等.离子色谱法同时测定牛乳及牛乳制品中硫氰酸盐和高氯酸盐[J].沈阳医药科技大学学报,2013,30(8):601-604.
[6]U.S.Food and Drug Administration.Revision 2:rapid determination of perchlorate anion in foods by ion chromatography-tandem mass spectrometry [EB/OL].(2005-04-12) [2017-04-01].ttps://www.fda.gov/Food/FoodborneIllnessContaminants/ChemicalContaminan-ts/ucm077793.htm.
[7]SHI Y L,ZHANG P,WANG Y W,et al.Perchlorate in sewage sludge,rice,bottled water and milk collected from different areas in China[J].Science Direct,2007,33(7):955-962.
[8]ARIBI H E,BLANC Y J C L,ANTONSEN S,et al.Analysis of perchlorate in foods and beverages by ion chromatography coupled with tandem mass spectrometry (IC-ESI-MS/MS)[J].Analytica Chimica Acta,2006,567(1):39-47.
[9]KIRK A B,KROLL M,DYKE J V,et al.Perchlorate,iodine supplements,iodized salt and breast milk iodine content[J].Science of the Total Environment,2012,420(1):73-78.
[10]BLASINI L V,BLOUNT B C,SANTOS S O,et al.Perchlorate exposure and dose estimates in infants[J].Environ Sci Technol,2011,45(9):4127-4132.
[11]BORJAN M,MARCELLA S,BLOUNT B,et al.Perchlorate exposure in lactating women in an urban community in New Jersey[J].Science of the Total Environment,2011,409(3):460-464.
[12]LI Y T,GEORGE E J.Analysis of perchlorate in water by reversed phase LC/ESI-MS/MS using an internal standard technique[J].Analytical Chemistry,2005,77(14):4453-4458.
[13]PISARENKO A N,STANFORD B D,QUINONES O,et al.Rapid analysis of perchlorate,chlorate and bromate ions in concentrated sodium hypochlorite solutions[J].Analytica Chimica Acta,2010,659(1/2):216-223.
[14]黄晓兰,罗辉泰,吴惠勤,等.奶粉中高氯酸盐的液相色谱-串联质谱测定[J].分析测试学报,2009,28(8):896-899.
[15]中华人民共和国国家质量监督检验检疫总局.进出口食品中高氯酸盐的测定 液相色谱-质谱/质谱法:SN/T 4089—2015[S].北京:中国标准出版社,2015.
[16]HOFF R B,RBENSAM G,JANK L,et al.Analytical quality assurance in veterinary drug residue analysis methods:matrix effects determination and monitoring for sulfonamides analysis[J].Talanta,2015,132(6):443-450.