 马妍,陈頔,陈曦,刘婷婷,李琴.分散固相萃取-分散液液微萃取-气相色谱-质谱联用法测定中草药类保健食品中29种有机磷农药残留[J].中国食品卫生杂志,2017,29(5):556-560.
马妍,陈頔,陈曦,刘婷婷,李琴.分散固相萃取-分散液液微萃取-气相色谱-质谱联用法测定中草药类保健食品中29种有机磷农药残留[J].中国食品卫生杂志,2017,29(5):556-560.DOi:10.13590/j.cjfh.2017.05.007
分散固相萃取-分散液液微萃取-气相色谱-质谱联用法测定中草药类保健食品中29种有机磷农药残留
(1.中国疾病预防控制中心营养与健康所 卫计委微量元素营养重点实验室,北京 100050; 2.中国环境科学研究院 环境基准与风险评估国家重点实验室,北京 100012; 3.中国环境科学研究院环境标准研究所,北京 100012)
作者简介:马妍 女 助理研究员 研究方向为营养与食品安全 E-mail:mayan_556@163.com
通信作者:李琴 女 研究员 研究方向为环境与健康 E-mail:liqin529luck@163.com
收稿日期:2017-06-23
基金项目:国家自然科学基金青年科学基金资助项目(31400097)
摘要:目的 建立测定中草药类保健食品中有机磷农药残留的分散固相萃取-分散液液微萃取-气相色谱-质谱联用法。方法 样品经含0.5%甲酸的乙腈超声提取,上清液经含无水硫酸镁、N-丙基乙二胺(PSA)、C18的分散固相萃取管(d-SPE)净化后,采用分散液液微萃取(DLLME)步骤,用氯仿萃取、浓缩目标化合物。结果 在最优条件下,线性范围为0.05~1.00 μg/ml,线性相关系数在0.999以上,检出限(LOD)为0.3~3.0 μg/kg,29种有机磷农药的平均加标回收率在70.3%~107.5%之间,相对标准偏差均<10%。结论 该方法具有简便快速、准确灵敏、萃取效率高等特点,可用于中草药类保健食品中有机磷农药残留的检测。
关键词:
分散固相萃取; 分散液液微萃取; 气相色谱-质谱; 有机磷农药; 保健食品; 中草药; 食品污染物; 测定
中图分类号: R155 文献标识码:A 文章编号:1004-8456(2017)05-0556-05
Determination of 29 organophosphorus pesticide residues in health food from Chinese herbal
medicines by dispersive solid phase extraction and dispersive liquid-liquid microextraction
coupled with gas chromatography-mass spectrometry
medicines by dispersive solid phase extraction and dispersive liquid-liquid microextraction
coupled with gas chromatography-mass spectrometry
(1.Key Laboratory of Trace Element Nutrition,National Institute for Nutrition and Health China CDC, Beijing 100050,China; 2.State Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences,Beijing 100012,China; 3.Environmental Standards Institute,Chinese Research Academy of Environmental Sciences,Beijing 100012,China)
Abstract:Objective To establish a method for the determination of pesticide residues in health food made from Chinese herbal medicines by dispersive solid phase extraction and dispersive liquid-liquid microextraction coupled with gas chromatography-mass spectrometry. Methods The samples were ultrasonic extracted by acetonitrile containing 0.5% formic acid. The supernatant was purified by dispersive solid phase extraction (d-SPE) extraction tube with anhydrous magnesium sulfate, N-propyl ethylenediamine (PSA) and C18, and then the target compounds were extracted and concentrated by the method of dispersive liquid-liquid microextraction (DLLME). Results Under optimal conditions, the linear range was 0.05-1.00 μg/ml, the linear correlation coefficient was above 0.999, the limit of detection was 0.3-3.0 μg/kg, the average recoveries of 29 kinds of pesticides were between 70.3% and 107.5%, and the relative standard deviation was less than 10%. Conclusion The method was simple, rapid, accurate and sensitive, and it could be used for the detection of organophosphorus pesticide residues in health food made from Chinese herbal medicines.
Key words:
Dispersive solid phase extraction; dispersive liquid-liquid microextraction; gas chromatography-mass spectrometry; organophosphorus pesticides; health food; Chinese herbal medicine; food contaminants; determination
欧盟食品安全委员会[1]、国际食品法典委员会(Codex标准)[2]和我国GB 2763—2016《食品安全国家标准 食品中农药最大残留限量》[3],严格规定了农牧产品中农药最大残留限量(maximum residue limits,MRLs),但是药用植物农药残留限量相关标准缺失严重。由于农药的广泛使用和长期使用,药用植物等原料中的农药残留不可避免。《中华人民共和国药典》2015版[4]中明确提出,农药残留是影响中药材安全性的主要风险因素之一。2015版药典对黄芪、西洋参、人参等6个中药品种制定了有机氯农药(organochloride pesticides,OCPs)残留限量标准,并增订了药材、饮片及制剂中部分农药多残留质谱测定法,包括气相色谱-串联质谱法同时测定74种农药残留,其中含11种有机磷农药,目前药典中尚无植物类中药有机磷农药残留规定或限量。中草药保健食品原材料主要以药食同源的中药材、植物提取物为主,一些加工过程,如浓缩、干燥、精油提炼等,可能会导致农药残留水平升高,还可能导致某些农药成分发生变化,产生毒性更高的代谢产物[5],因此,为了避免中草药保健食品中农药超标、农药联合毒性对人体造成伤害,加强中草药保健食品农药残留监管及风险评估的力度,建立一套快速、简便、灵敏的检测方法十分必要。 目前,农药残留的前处理方法主要有液液萃取法[6]、QuEChERS法[7]、固相萃取法[8]、基质固相分散法[9]等,其中分散液液微萃取(DLLME)法是近年来发展迅速的样品前处理技术,具有操作简便、富集倍数高、快速,尤其是有机溶剂用量少等优点,已经逐步在农药残留领域应用[10-12]。在DLLME法萃取程序中,微量萃取剂在分散剂作用下与水溶液形成三相乳浊液体系,离心后萃取剂集中到最下层。在三相分配平衡过程中,萃取剂形成了细小的分散液滴,增大了与分析物的接触面积,能够快速有效达到分配平衡,完成萃取。本试验以中草药保健食品常见的3种剂型(片剂、硬胶囊剂和液体制剂)作为研究基质,采用分散固相萃取(d-SPE)和DLLME法联用的前处理技术,结合气相色谱-质谱(GC-MS)法同时测定了中草药类保健食品中29种农药残留。
1 材料与方法
1.1 材料
1.1.1 样品来源
15份中草药保健食品均购自北京市药店,其中液体制剂(口服液)5份,固体制剂(含片剂、硬胶囊剂)10份。
1.1.2 主要仪器与试剂
Thermo 1300-TSQ8000 Evo 气相色谱-质谱联用仪(美国Thermo)、离心机、涡旋振荡器、氮吹仪、电热恒温水浴锅、干式恒温混匀仪、Milli-Q Reference型超纯水机、商品化12 ml PSA/C18净化管[美国色谱科,填料为150 mg N-丙基乙二胺(PSA)、150 mg C18、900 mg无水硫酸镁]。 29种农药对照品均购自德国Dr. Ehrensotrofer,标准品用丙酮配制成1 000 μg/ml的储备液,于4 ℃冰箱中避光保存。四氯化碳、氯苯、氯仿、乙腈、丙酮、氯化钠、无水硫酸钠均为分析纯,试验用水为超纯水。
1.2 方法
1.2.1 标准溶液的配制
分别吸取29种农药1 000 μg/ml的储备液1.00 ml置于同一50 ml容量瓶中,配制成质量浓度为20.0 μg/ml的混合标准液。之后用不含有目标化合物的样品溶液稀释成质量浓度为1.0 μg/ml的混合标准液,并逐步配制成0.00、0.05、0.10、0.25、0.50、1.00 μg/ml的标准曲线系列溶液。
1.2.2 样品前处理
片剂、硬胶囊剂保健食品提取:称取粉碎样品2.0 g(片剂去外层辅料、胶囊去壳),置于50 ml离心管中,加入5 ml水和10 ml含0.5%甲酸的乙腈溶液,涡旋混合2 min,在混匀仪上振摇15 min后,加入NaCl约2.5 g,继续振摇5 min,8 000 r/min离心5 min。转移上清液至d-SPE管。 液体制剂保健食品提取:称取10.0 g样品溶液,置于50 ml离心管中,加入10 ml含0.5%甲酸的乙腈溶液,涡旋混合2 min,在混匀仪上振摇15 min后,加入NaCl约3.5 g,继续振摇5 min,8 000 r/min离心5 min。转移上清液至d-SPE管。 d-SPE净化:取6.5 ml上清液加到商品化12 ml PSA/C18净化管中净化,涡旋振荡混合1 min,8 000 r/min离心5 min。取净化好上清液5 ml到10 ml螺口锥型离心管中,室温水浴通氮气吹至约余半滴大小,在通风橱内将余下的溶剂挥发至近干。 DLLME法萃取:向含有浓缩样品的10 ml离心管中加入5 ml 28 ℃超纯水,涡旋振荡30 s后,用微量注射器在离心管底部迅速注入氯仿52 μl、丙酮88 μl,之后28 ℃恒温混匀2 min,形成均匀的乳浊液,于8 000 r/min离心5 min。用微量注射器将沉淀相转移至含150 μl锥形微量内插管的样品瓶中,供GC-MS分析检测。
1.2.3 仪器条件
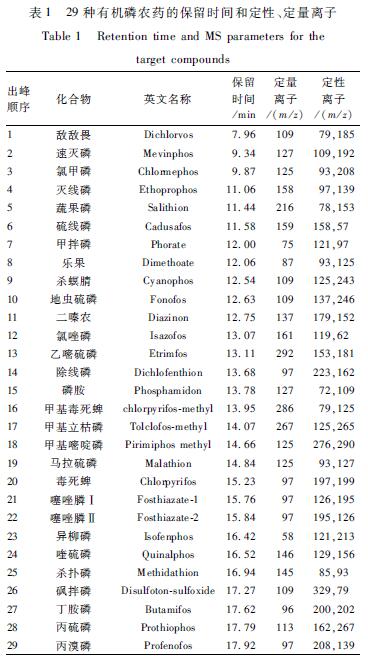
色谱:毛细管色谱柱TG-5MS(30 m×0.25 mm,0.25 μm)配置5 m脱活保护柱;高纯氦(99.999%),流速梯度1.2 ml/min保持30 min,之后3 ml/min保持7.2 min;进样模式:程序升温气化(PTV)进样口,不分流反吹进样,不分流时间2.0 min,吹扫流量5.0 ml/min;传输压力:210 kPa;进样口初始温度75 ℃,进样后以2.5 ℃/s快速升至300 ℃,保持3.0 min,进样净化阶段再以14.5 ℃/s快速升至330 ℃,保持20.0 min;色谱柱升温程序:90 ℃保持5 min,然后以25 ℃/min升至180 ℃,以5 ℃/min升至280 ℃,再以10 ℃/min升至300 ℃,保持5 min。进样体积1 μl。 质谱:传输线温度250 ℃,离子源温度300 ℃;电离方式:电子轰击电离源(EI);电离能量70 eV;溶剂延迟6.5 min。保留时间和选择离子见表1。
|
表1 29种有机磷农药的保留时间和定性、定量离子 Table 1 Retention time and MS parameters for the target compounds |
2 结果与分析
2.1 DLLME法萃取条件优化
2.1.1 分散剂与萃取剂的选择
选择合适的萃取溶剂是影响DLLME法萃取效果的重要因素之一,萃取溶剂应与目标化合物满足极性“相似相容”原理,同时对样品中干扰物质的溶解度尽量低,减少基质效应。萃取溶剂还应选择不溶于水且密度大于水的溶剂,在离心后能沉积到下层,如一些氯代烃类。本试验采用经d-SPE净化的保健食品样品水溶液5 ml,添加混合标准样品至浓度为0.5 μg/ml,分别对50 μl不同萃取剂四氯化碳、氯苯、氯仿进行回收率考察;并用0.5 μg/ml的溶剂混合标准样品对基质效应进行了考察。结果表明,氯仿具有最好的萃取效果,回收率较高且稳定,色谱峰形好;四氯化碳的萃取效率最差;采用氯苯萃取时,磷胺极性较强农药回收率偏低。 分散剂要求既溶于水又溶于萃取剂,能与水、萃取剂构成微小液滴的三相乳化体系。试验比较了乙腈与丙酮的分散效果,发现两者均可以与氯仿、水溶液形成均匀的乳化体系,丙酮做分散剂时氯仿萃取液中的基质效应较小。这可能是由于丙酮与氯仿极性更相近,转移了氯仿中的部分杂质。本试验最终选择丙酮作分散剂,将d-SPE与DLLME法有效结合,使DLLME法可以应用于基质复杂样品前处理中。
2.1.2 氯化钠用量、混匀时间、pH的选择
氯化钠常用来调节样品溶液的离子强度,从而影响待测物在水相中的溶解度。本试验考察了DLLME法萃取程序中氯化钠用量分别为0.3~1.5 g时对保健食品农药残留平均回收率的影响,试验结果表明,若增加氯化钠浓度,富集倍数和回收率均明显降低。显示出随着离子强度的增加,水与有机溶剂互溶减小,难以形成均匀微小液滴的三相乳浊体系,使得沉积相体积增加、萃取效率降低,因此本试验选择不加氯化钠。 考察了恒温时混匀萃取平衡时间分别为1.0~5.0 min时对萃取效率的影响。结果表明,当混匀时间>1.5 min时,萃取效率达到稳定状态,最终选择混匀时间为2 min。 在提取时加入酸化乙腈,可以增加目标化合物在乙腈中的稳定性,提高提取率。弱酸性环境也会增加一些带酸性基团(如羧基、酰胺基、醚基等)有机磷农药在乙腈中的溶解度。pH降低时,电离平衡向中性分子方向移动,而中性分子比离子化分子在有机相中的溶解性更好,更容易转移到萃取剂中,萃取率相对更高。为了选择合适的样品溶液pH值,本试验考察了加入不同酸度乙腈(分别含0%甲酸、0.5%甲酸、1%甲酸)时的萃取率。结果表明,使用含0.5%甲酸的乙腈提取保健食品样品时,获得的萃取率最高。
2.1.3 DLLME法萃取的响应面试验设计、模型及优化方案
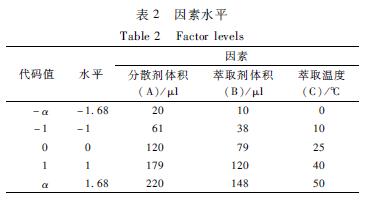
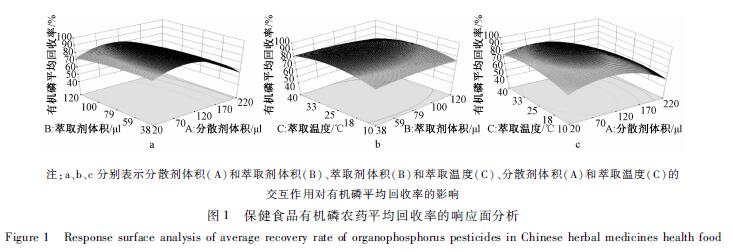
根据单因素试验结果,本试验采用Design-Expert 8.0.6软件对选择分散剂体积(A)、萃取剂体积(B)和萃取温度(C)等因素进行响应面(response surface)优化,以保健食品中农药的平均回收率为评价指标,建立高效的试验模型。按照中心组合设计(central composite design)原理,每因素设5水平,分别用代码值-α、-1、0、1、α表示,因素水平见表2。得到的拟合方程差异有统计学意义(r=0.91,P<0.05),因素A、B、C对综合评分(平均回收率)的影响见效应面图1。由此可知,在3个因素的互相作用下,分散剂丙酮对综合萃取效率的影响较大,回收率随着分散剂丙酮的用量增大而增大,但在达到88 μl之后,若继续提高参数,回收率反而下降。温度对萃取效率的影响基本呈二次函数关系,在27.0~29.0 ℃时达到最大。萃取剂体积主要影响化合物的富集倍数,体积越小萃取效率越高,但体积过小时,基质效应较高、重现性变差,且不适于分析仪器的自动进样。结合实际应用,最终确定萃取优化条件为温度28 ℃、分散剂丙酮体积88 μl、萃取剂氯仿体积52 μl。
|
表2 因素水平 Table 2 Factor levels 注:a、b、c分别表示分散剂体积(A)和萃取剂体积(B)、萃取剂体积(B)和萃取温度(C)、分散剂体积(A)和萃取温度(C)的 交互作用对有机磷平均回收率的影响 |
|
图1 保健食品有机磷农药平均回收率的响应面分析 Figure 1 Response surface analysis of average recovery rate of organophosphorus pesticides in Chinese herbal medicines health food |
2.2 方法的线性关系
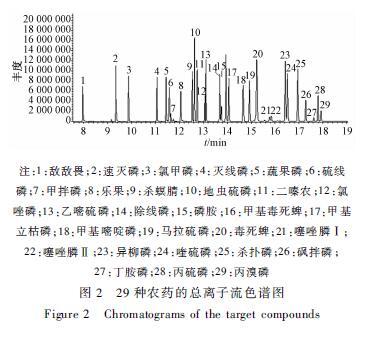
将1.2.1配制的农药标准曲线系列溶液在1.2.3仪器条件下进样,分别测定其峰面积,并以此作为纵坐标,农药标准液的质量浓度为横坐标,绘制标准曲线。结果表明,29种农药在0.05~1.00 μg/ml内具有良好的线性关系,其相关系数均>0.999,样品添加浓度为0.5 μg/ml水平的29种农药总离子流色谱图见图2。
|
注:1:敌敌畏;2:速灭磷;3:氯甲磷;4:灭线磷;5:蔬果磷;6:硫线 磷;7:甲拌磷;8:乐果;9:杀螟腈;10:地虫硫磷;11:二嗪农;12:氯 唑磷;13:乙嘧硫磷;14:除线磷;15:磷胺;16:甲基毒死蜱;17:甲基 立枯磷;18:甲基嘧啶磷;19:马拉硫磷;20:毒死蜱;21:噻唑膦Ⅰ; 22:噻唑膦Ⅱ;23:异柳磷;24:喹硫磷;25:杀扑磷;26:砜拌磷; 27:丁胺磷;28:丙硫磷;29:丙溴磷 图2 29种农药的总离子流色谱图 Figure 2 Chromatograms of the target compounds |
2.3 方法的回收率和精密度
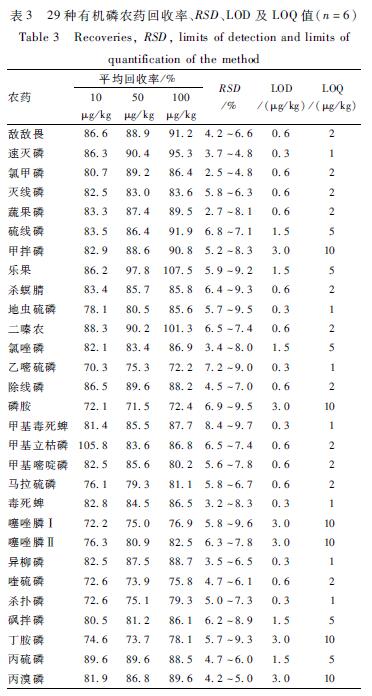
在经分析不含上述29种农药的保健食品中,分别进行添加水平为10、50和100 μg/kg的加标回收试验,每个浓度均做6个平行样,29种农药的平均回收率为70.3%~107.5%,相对标准偏差(RSD)均<10%,符合农药残留检测的要求,具体数据见表3。
2.4 方法的检出限与定量限
本方法各农药检出限(LOD)是采用检测器3倍基线噪声,结合所用农药标准溶液的浓度、定容体积、稀释倍数、称样量和响应值(峰高)各因素计算所得,范围为0.3~3.0 μg/kg,LOD与定量限(LOQ)见表3,均低于我国相关标准规定的蔬菜农药MRLs值的要求。
2.5 实际保健食品样品分析
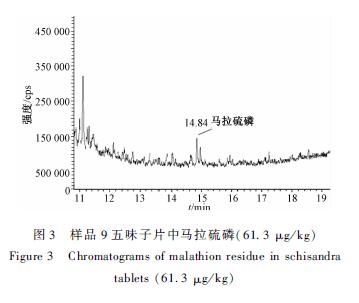
15份保健食品按1.2.2样品前处理方法平行制备2份,按1.2.3仪器条件进行测定。结果显示,样品1奶蓟草胶囊中毒死蜱的检出量是88.6 μg/kg、样品6丹参胶囊中毒死蜱的检出量是97.2 μg/kg。图3为样品9五味子片中含马拉硫磷,其含量为61.3 μg/kg。
|
表3 29种有机磷农药回收率、RSD、LOD及LOQ值(n=6) Table 3 Recoveries, RSD, limits of detection and limits of quantification of the method |
|
图3 样品9五味子片中马拉硫磷(61.3 μg/kg) Figure 3 Chromatograms of malathion residue in schisandra tablets (61.3 μg/kg) |
3 小结
本试验建立了d-SPE-DLLME-GC-MS法测定中 草药保健食品有机磷农药残留,可实现29种农药残留检测。该法前处理过程简单,减少了基质效应的影响,提高了检测速度,方法的线性范围、准确度、精密度及检出限均满足相关标准的要求,适合农药检测任务量日益增加的实际需要。
参考文献
[1] EUROPEAN COMMISSION. Maximum Residue Levels, pestic-ides database redirection[DB/OL].(2017-01-25)[2017-03-02]. http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database-redirect/index_en.htm.
[2] FAO. Codex maximum limits for pesticide residues, pesticide residues in food and feed[DB/OL].(2016-08-18)[2017-03-02]. http://www.fao.org/fao-who-codexalimentarius/stan-dards/pestres/zh/.
[3] 中华人民共和国国家卫生和计划生育委员会,中华人民共和国农业部,国家食品药品监督管理总局.食品安全国家标准 食品中农药最大残留限量:GB 2763—2016[S]. 北京:中国标准出版社,2016.
[4] 国家药典委员会.中华人民共和国药典[M]. 北京:中国医药科技出版社,2015:209-223.
[5] 李云成,孟凡冰,陈卫军. 加工过程对食品中农药残留的影响[J].食品科学,2012,33(5):315-317.
[6] 于桂芳,王宪平,胡军华,等. GC-ECD法测定5种保健食品中六六六和滴滴涕的农药残留[J]. 药学与临床研究,2015,23(5):468-470.
[7] ANASTASSIADES M,LEHOTAY S J,STAJNBAHER D,et al. Fast and easy multi-residue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction" for the determination of pesticide residues in produce[J]. Journal of AOAC International,2003,86(2):412-431.
[8] 陈菲菲,李星芝,王俊全,等. 一种同时测定保健食品中多种农药残留的方法[J]. 食品研究与开发,2012,33(10):123-126.
[9] AQUINO A,WANDERLEY K A,PAIVA-SANTOS C O,et al. Coordination polymer adsorbent for matrix solid-phase dispersion extraction of pesticides during analysis of dehydrated Hyptis pectinata medicinal plant by GC/MS[J]. Talanta,2010,83(2):631-636.
[10] WANG H Z, HU L, LI W Z,et al. A rapid and simple pretreatment method for benzoylurea insecticides in honey samples using in-syringe dispersive liquid-liquid microextraction based on the direct solidification of ionic liquids[J]. Journal of Chromatography A,2016,1471(11):60-67.
[11] CHEN B,WU F Q,WU W D,et al. Determination of 27 pesticides in wine by dispersive liquid-liquid microextraction and gas chromatography-mass spectrometry [J]. Microchemical Journal,2016,126(5):415-422.
[12] 崔世勇,樊珠凤,姜丽华. 悬浮固化分散液液微萃取-气相色谱-质谱法测定水中10种有机磷农药[J].现代预防医学,2016,43(11):2048-2066.
[2] FAO. Codex maximum limits for pesticide residues, pesticide residues in food and feed[DB/OL].(2016-08-18)[2017-03-02]. http://www.fao.org/fao-who-codexalimentarius/stan-dards/pestres/zh/.
[3] 中华人民共和国国家卫生和计划生育委员会,中华人民共和国农业部,国家食品药品监督管理总局.食品安全国家标准 食品中农药最大残留限量:GB 2763—2016[S]. 北京:中国标准出版社,2016.
[4] 国家药典委员会.中华人民共和国药典[M]. 北京:中国医药科技出版社,2015:209-223.
[5] 李云成,孟凡冰,陈卫军. 加工过程对食品中农药残留的影响[J].食品科学,2012,33(5):315-317.
[6] 于桂芳,王宪平,胡军华,等. GC-ECD法测定5种保健食品中六六六和滴滴涕的农药残留[J]. 药学与临床研究,2015,23(5):468-470.
[7] ANASTASSIADES M,LEHOTAY S J,STAJNBAHER D,et al. Fast and easy multi-residue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction" for the determination of pesticide residues in produce[J]. Journal of AOAC International,2003,86(2):412-431.
[8] 陈菲菲,李星芝,王俊全,等. 一种同时测定保健食品中多种农药残留的方法[J]. 食品研究与开发,2012,33(10):123-126.
[9] AQUINO A,WANDERLEY K A,PAIVA-SANTOS C O,et al. Coordination polymer adsorbent for matrix solid-phase dispersion extraction of pesticides during analysis of dehydrated Hyptis pectinata medicinal plant by GC/MS[J]. Talanta,2010,83(2):631-636.
[10] WANG H Z, HU L, LI W Z,et al. A rapid and simple pretreatment method for benzoylurea insecticides in honey samples using in-syringe dispersive liquid-liquid microextraction based on the direct solidification of ionic liquids[J]. Journal of Chromatography A,2016,1471(11):60-67.
[11] CHEN B,WU F Q,WU W D,et al. Determination of 27 pesticides in wine by dispersive liquid-liquid microextraction and gas chromatography-mass spectrometry [J]. Microchemical Journal,2016,126(5):415-422.
[12] 崔世勇,樊珠凤,姜丽华. 悬浮固化分散液液微萃取-气相色谱-质谱法测定水中10种有机磷农药[J].现代预防医学,2016,43(11):2048-2066.